
When Your Prostate Cancer Drug Becomes Cancer Fuel - 072

Prostate cancer is a trickster
Prostate cancer has many tricks that most men don’t know about, and this one is a doozy. It doesn’t just stop responding to drugs. Sometimes it learns to use the drugs as a catalyst for growth.
There is a well-documented molecular mechanism in which specific mutations in the prostate cancer cell cause a drug designed to block cancer growth to start fueling it instead. This ability to flip the drug from an antagonist (blocker) to an agonist (activator) is called the antagonist-to-agonist switch.
When the drug flips from blocker to activator, the cancer keeps growing, the PSA keeps rising, and neither the patient nor the doctor necessarily understands why. That’s because the mutation driving this flip is usually undetected unless the doctor is savvy enough to know it probably occurred or to test for it.
The androgen receptor is the lock that controls everything
At its foundation, prostate cancer is driven by hormones. Most of these cancer cells contain a protein called the androgen receptor (AR).
Think of the AR as a gate that swings open when testosterone or another androgen fits into the receptor. Once in the cell, these androgens act as fuel, stimulating the cancer to grow, divide, and spread.
Most drugs used to treat prostate cancer attempt to keep that gate shut or reduce the flow of fuel through it. Androgen deprivation therapy drugs like Lupron starve the system by reducing testosterone in the blood.
A newer, more powerful class called androgen receptor pathway inhibitors (ARPIs) addresses the problem from multiple angles. Some block the receptor directly so testosterone can’t bind to it, while others cut off the tumor’s ability to manufacture its own androgens.
ARPIs include:
enzalutamide (Xtandi)
apalutamide (Erleada)
darolutamide (Nubeqa)
abiraterone acetate (Zytiga, Yonsa)
Older first-generation drugs called antiandrogens, such as bicalutamide, work similarly to ARPIs but with less potency and more vulnerability to resistance.
These drugs work at first, but the pressures they exert at the molecular level cause these very smart cancer cells to figure out how to survive.
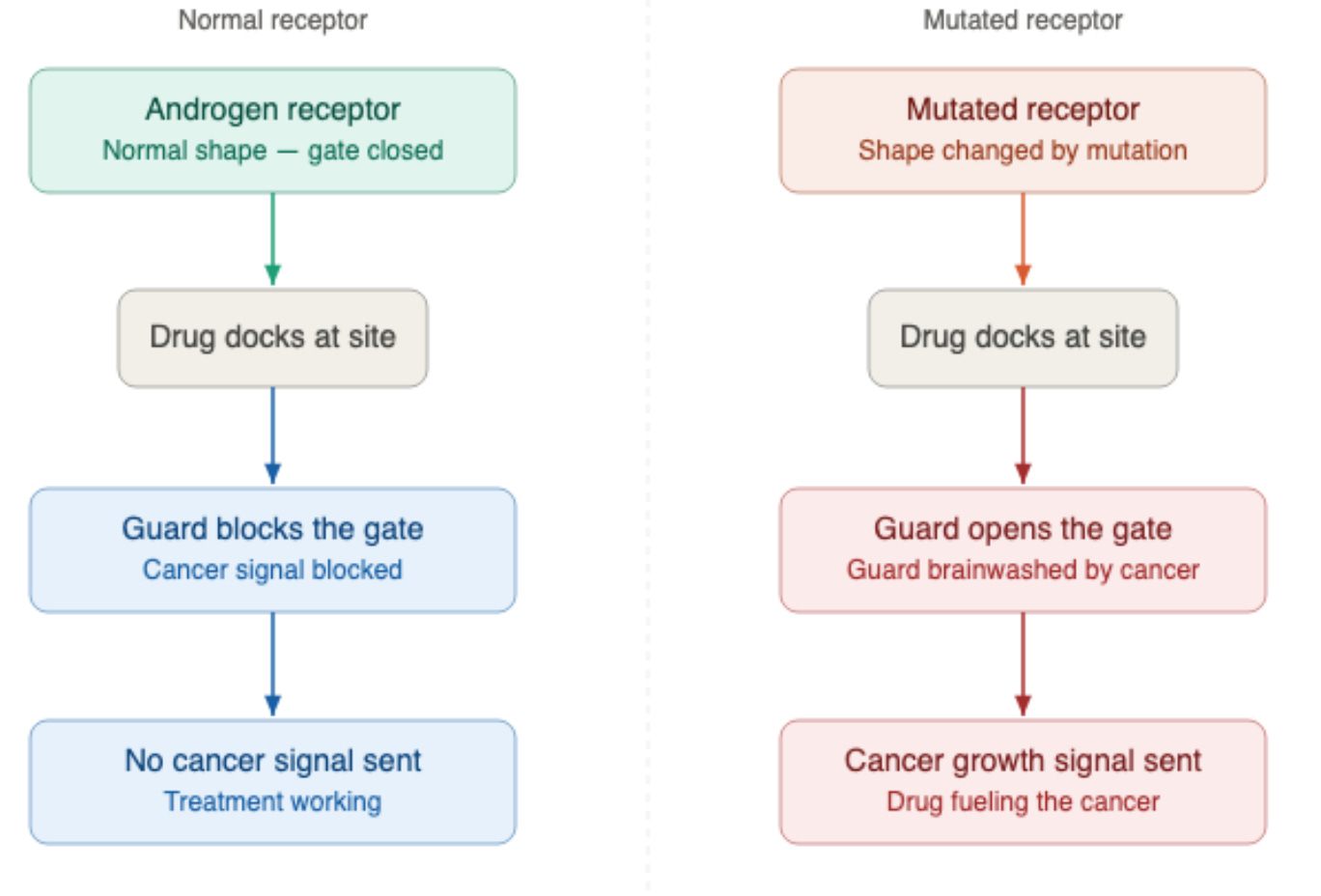
How the drug becomes the enemy’s recruit
Over time, under the constant pressure of the drug, the cancer mutates. It causes mutations in the androgen receptor’s ligand-binding domain, the region where drugs and hormones bind. This specific type of mutation creates an error in the genetic code and is called a point mutation in which one amino acid in the receptor is swapped for a different one.
This amino acid swap reshapes the docking site just enough to flip the drug’s entire function, creating this antagonist-to-agonist switch.
Think of the drug as a security guard you hired to stand at the front gate to keep intruders out. The mutation is like the cancer brainwashing that guard. He still stands at the gate, but instead of blocking the intruders, he opens the door and waves them in.
The specific drugs and what happens to them
Not every drug triggers the same mutation, and not every mutation affects every drug the same way. Here is what the research shows:
Bicalutamide and the W742 mutations
Bicalutamide is an older first-generation antiandrogen used for decades. Mutations at position 742 of the receptor, specifically W742C and W742L, cause bicalutamide to activate the receptor rather than block it.
In patients who develop these mutations, bicalutamide is literally feeding the cancer, causing the PSA to rise. If the clinician knows the potential for this to occur and stops the drug, one might see a significant PSA drop. This phenomenon is called the bicalutamide withdrawal response.¹
Bicalutamide monotherapy is rarely used in the United States today, where it is more commonly added to ADT as combination therapy or used in other specific clinical contexts. This withdrawal response is more common in countries where clinicians use it as monotherapy.
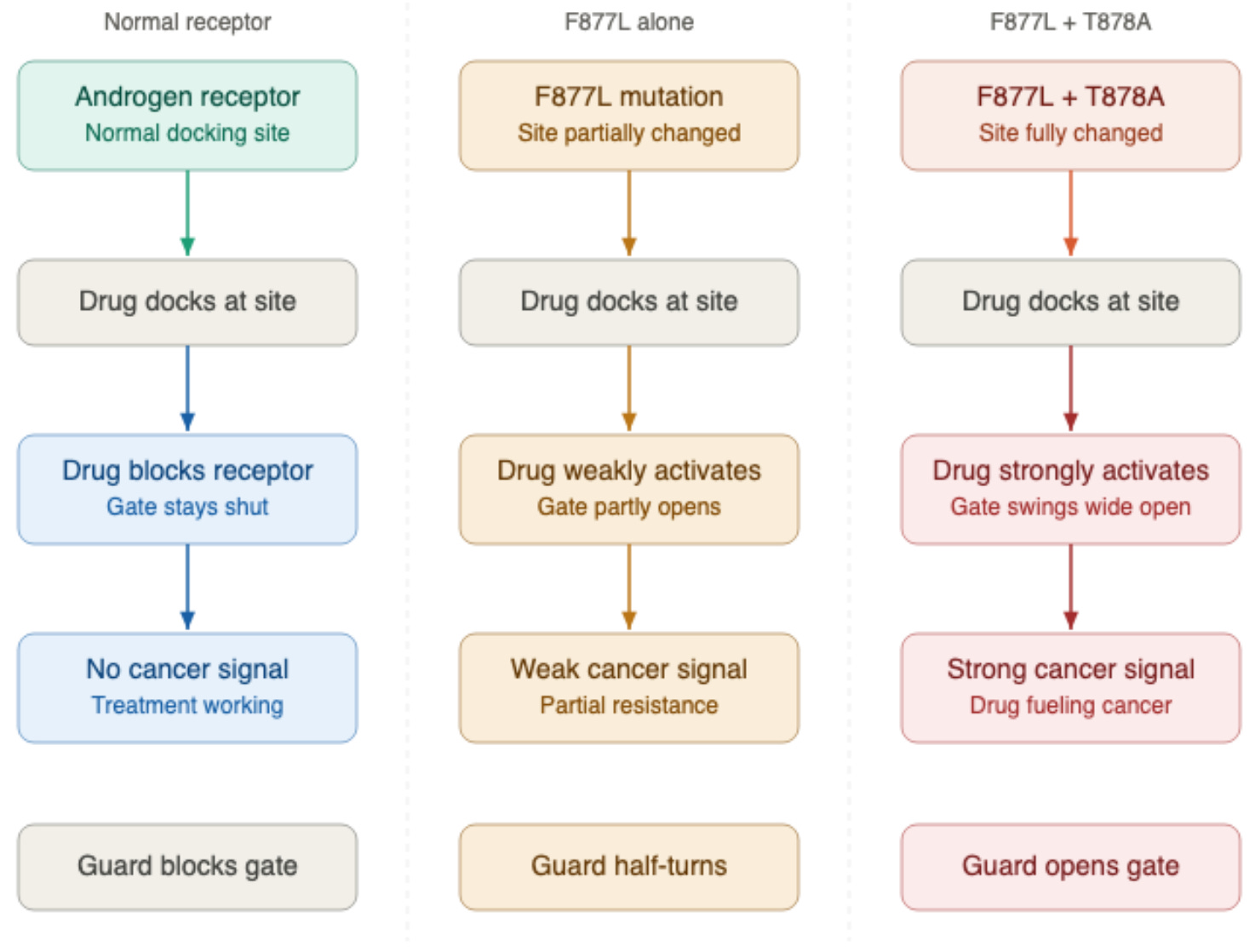
Enzalutamide and F877L
Enzalutamide is a far more potent second-generation drug, designed to overcome some of the resistance mechanisms that defeated older drugs like bicalutamide. But the cancer can still find a way to overcome it.
The F877L mutation, involving an amino acid substitution at position 877, reshapes the receptor’s docking site so that enzalutamide now acts as a partial agonist rather than a blocker.² The same mutation has been documented with apalutamide (Erleada), where F877L similarly converts the drug from a blocker into an activator, detected in approximately 3.7% of patients who progressed on apalutamide in a prospective trial.¹⁶
The agonist activity of F877L alone is weak to moderate. When F877L occurs alongside a second mutation called T878A, the agonist activity becomes substantially stronger — an effect that has been studied specifically with enzalutamide.² Whether the same compounding effect applies equally to apalutamide is not yet known.
Abiraterone’s two unique traps
Abiraterone works differently from the antiandrogens above. Rather than blocking the androgen receptor directly, it blocks an enzyme called CYP17A1 that the body uses to produce testosterone and other androgens.
Abiraterone is effective enough to extend survival significantly in men with metastatic castration-resistant prostate cancer, but it has two separate trap doors built into the way it works.
The first is the prednisone trap.
When abiraterone blocks CYP17A1, cortisol production falls. The body responds by releasing a surge of ACTH, a hormone that signals the adrenal glands to produce more steroids. That surge drives overproduction of the compounds deoxycorticosterone and corticosterone, which accumulate upstream of the CYP17A1 block.
These compounds act like mineralocorticoids, promoting sodium absorption and fluid retention, which raises blood pressure and lowers potassium.⁴ Prednisone or prednisolone is given alongside abiraterone specifically to suppress that ACTH surge and blunt this chain reaction.
The problem arises when the L702H mutation develops, involving an amino acid substitution at position 702 that alters the receptor’s binding pocket and makes it sensitive to glucocorticoids, including prednisone and the body’s own cortisol.
In men with the L702H mutation, the prednisone that must accompany abiraterone directly activates the androgen receptor and drives cancer growth. The drug given to manage abiraterone’s side effects becomes the cancer’s new fuel source.
L702H has been detected in approximately 10% to 15% of patients after abiraterone treatment, though rates vary by detection method and patient population.³
The second trap involves progesterone.
When abiraterone blocks CYP17A1, hormone precursors such as progesterone, which would normally be converted to androgens, start to back up, causing progesterone levels to rise dramatically, up to 50-fold above normal.⁴
Normally, that wouldn’t matter much because a healthy androgen receptor isn’t sensitive to progesterone. But the T878A mutation changes that, making the receptor responsive to progesterone and other steroid hormones it would normally ignore, such as estradiol.
The T878A mutation, which substitutes an amino acid at position 878, creates a promiscuous receptor that is now activated by a much wider range of hormones, including progesterone. This mutation occurs in approximately 21% of patients after abiraterone treatment.⁴
There is also a third, less discussed mechanism. The body metabolizes abiraterone to 5α-abiraterone, which can act as a direct androgen receptor agonist independent of any mutations.⁵
A rant from me
On a side note, these occurrences with abiraterone are on my list of several reasons why I don’t like this drug. It is a dirty drug, meaning it has a slew of serious side effects and mutation potentials, and requires that you take a corticosteroid, which has its own list of serious side effects.
For example, the LATITUDE clinical trial showed that 37% of men with mCSPC taking abiraterone developed hypertension, with 20% being severe (Grade 3).¹⁷ Even when researchers attempt to reduce the cost burden to patients by showing efficacy with a lower 250 mg dose taken with a low-fat meal, hypertension as a side effect was prevalent.
A case series from the University of Washington and Fred Hutchinson Cancer Center, though only a small case series and not validated in prospective studies, found that 76.9% of patients receiving this low-dose regimen developed high blood pressure during treatment.¹⁸ While these cases were manageable with additional medication, that incidence rate illustrates the high cardiovascular burden abiraterone carries compared to ARPIs like darolutamide.
The table below summarizes each mutation, the drug responsible, what it does, and how rarely it appears before treatment begins.

Why these mutations happen
These mutations are rarely found before treatment but frequently emerge as a response to the pressure of therapy. In treatment-naive patients, meaning those who have not yet received hormonal therapy, AR ligand-binding domain mutations are essentially undetectable in tumor tissue and circulating tumor DNA.¹⁴
The mutations described in this article are, in the overwhelming majority of cases, a direct consequence of treatment itself. The drugs create the very mutations that defeat them.
There is one narrow exception worth knowing. The T878A mutation, which emerges primarily under abiraterone treatment, has been detected in approximately 3% of men who had already received prior hormonal therapy before starting their first ARPI.¹⁵ But this still occurs in men exposed to hormonal therapy, whether it is ADT or abiraterone.
For all practical purposes, these mutations are created by the drugs used to treat this disease, which is precisely what makes them so clinically important to detect.
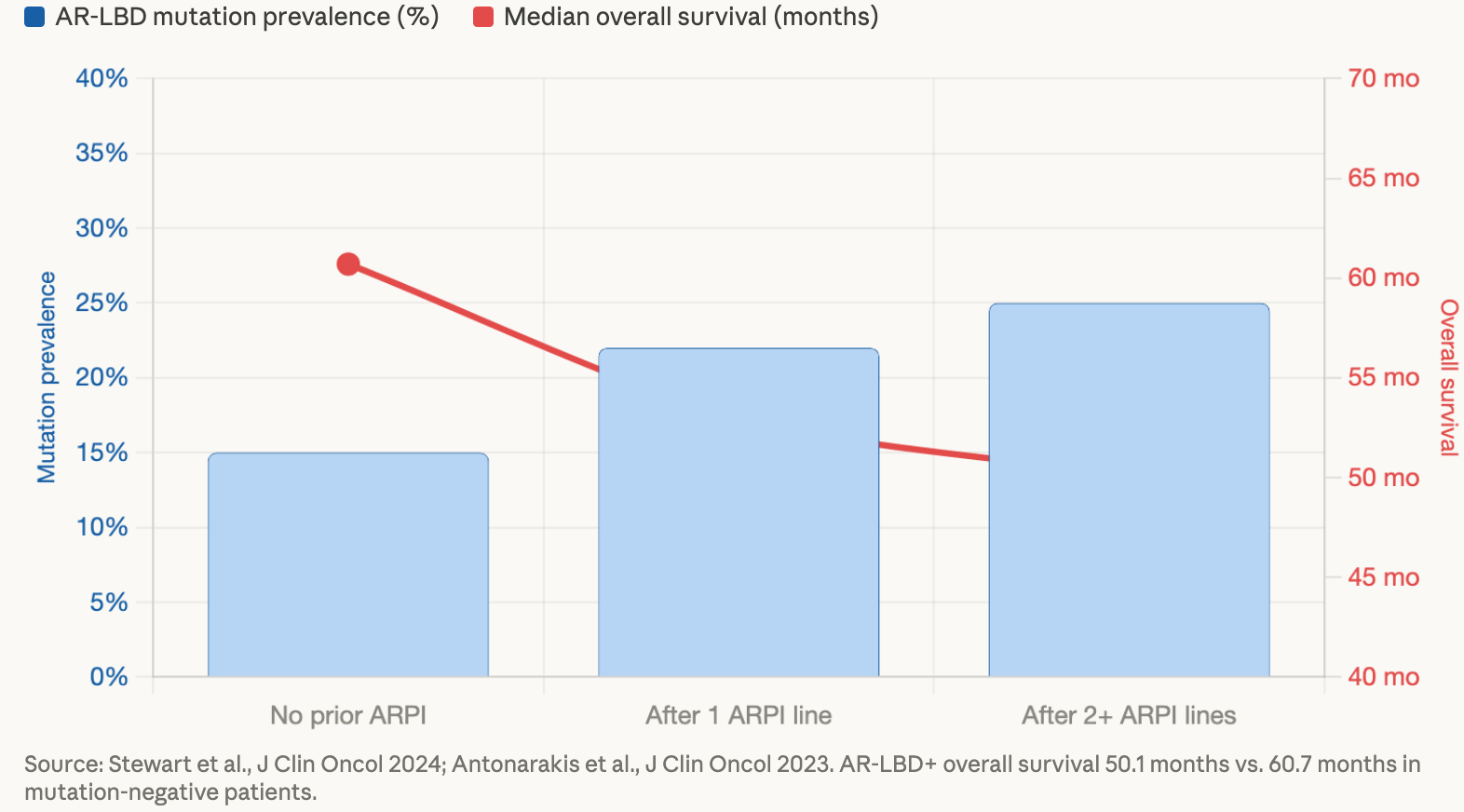
The data make this pattern visible. As men progress through more lines of therapy, the prevalence of these mutations climbs — and as that prevalence climbs, survival shortens.
When the clinical picture becomes a maze
These mutations don’t announce themselves. Without specific molecular testing, neither you nor your doctor can see them. What you can see is a rising PSA in the presence of an agonist-producing mutation, which can lead to a series of clinical decisions that make things worse rather than better.
Scenario 1 - The ARPI switch that backfires
A man is on enzalutamide, and his PSA starts rising. His doctor interprets this, reasonably, as the drug failing and considers switching to abiraterone, the other major ARPI available.
This is where knowing the mutation status becomes critical, because the outcome of that switch depends entirely on what is driving the rise in PSA.
If the reason enzalutamide stopped working is the F877L mutation, switching to abiraterone works through a different mechanism and isn’t directly affected by that mutation, so there may be a chance of a response.
But if the reason enzalutamide stopped working is L702H, switching to abiraterone introduces mandatory prednisone administration to a patient whose cancer is already primed to feed on glucocorticoids.
The doctor made a standard clinical consideration and inadvertently created conditions that allowed an accelerant to be poured on the fire.
Even when none of the agonist-switch mutations described in this article are present, switching one ARPI to another often fails for a separate reason entirely. Examples of ARPI switching classically involve switching a man failing enzalutamide to abiraterone acetate, or vice versa.
Prostate cancer can develop variants of the androgen receptor (AR) called AR splice variants that lack the docking site where all ARPIs bind. The most common of these variants is AR-V7.
A receptor lacking a docking site cannot be blocked or activated by any drug in this class, making it invisible to all ARPIs, regardless of which ARPI the doctor tries next.
This is one of the primary reasons the major oncology guidelines have become increasingly cautious about ARPI switching as a strategy. I have previously written about this issue as a design flaw in clinical trials that allow ARPI switching in control arms.
Given the generally low efficacy of an ARP switch, its use in clinical trials in a control arm is a disservice to men in the control arms and may make the study drug appear better than it is. Men in control arms of prostate cancer trials should be receiving the best of care options, which is not what they get with an ARPI switch.
The NCCN guidelines list switching from one ARPI to another as an option only for select patients who are not candidates for other recommended therapies, and it is conspicuously absent from the preferred and other recommended options in their post-ARPI treatment algorithm.⁶
The preferred recommendation after ARPI progression is docetaxel chemotherapy.⁶ The 2025 ASCO guideline on metastatic CRPC similarly does not recommend ARPI switching as a standard approach and instead directs clinicians toward docetaxel for patients who have received prior ARPI.⁷
The PLATO trial examining ARPI sequencing found that switching from abiraterone to enzalutamide yielded only a 27% PSA response rate and a median PSA progression-free survival of 5.7 months. Switching from enzalutamide to abiraterone was even worse, producing a 4% PSA response rate and just 1.7 months to PSA progression.⁸
The trial authors concluded that other available therapies, including taxane chemotherapy, should be strongly considered for patients who progress after first-line ARPI.⁸
Scenario 2 - The contaminated signal
A man on bicalutamide with a W742 mutation experiences PSA progression. His doctor stops bicalutamide and immediately starts the next drug. The PSA subsequently drops.
Two things are now happening simultaneously. Removing bicalutamide triggers a withdrawal response, where the W742-driven agonist activity disappears, and the PSA falls on its own. The new drug may or may not be contributing to that drop. With both changes happening at once, it becomes impossible to tell what’s actually causing the PSA to fall.
A brief drug holiday before starting the next therapy would have separated these two effects and given a much cleaner picture of what the cancer is actually responding to.
Scenario 3 - The L702H trap
The L7202H mutation is the most extensively documented in the peer-reviewed literature, and the one most likely to trap a patient in a cycle of ineffective sequential therapy.
L702H emerges primarily during abiraterone treatment because prednisone, which must be co-administered, activates the mutated receptor directly. But L702H does not disappear when a patient switches from abiraterone to enzalutamide.
A large real-world study detected L702H in 18% of patients treated with abiraterone alone, in 32% of patients treated with enzalutamide alone, and in 25% of patients treated with both drugs.⁹
The problem is that in real-world clinical practice, many patients continue glucocorticoids after switching from abiraterone to enzalutamide, for pain management, fatigue, or because clinicians are wary about abrupt steroid withdrawal after prolonged abiraterone use.
As long as glucocorticoids are circulating, L702H-positive clones remain activated. The cancer continues to grow, the PSA continues to rise, and the new drug gets blamed when the actual culprit is the steroid that nobody stopped.
Pearl: If glucocorticoids genuinely need to continue during enzalutamide therapy for legitimate symptom management, some clinicians prefer switching from prednisone to dexamethasone. Based on evidence that dexamethasone has a lower affinity for the L702H-mutated receptor than prednisone does.¹⁰
Scenario 4 - The prednisone trap compounded
A man on abiraterone develops L702H. His prednisone is now activating his cancer, his PSA is rising steadily, and it looks clinically like he is failing abiraterone. His doctor stops both abiraterone and prednisone and starts a new drug.
The PSA drops. But the drop may not indicate that the new drug is working. It may simply be that removing prednisone cuts off the fuel supply to the L702H clone. The new treatment gets credit it may not deserve, and weeks or months later, the PSA climbs again. At no point did anyone test for L702H.
A structurally different approach
Darolutamide (Nubeqa) is the newest ARPI drug, and its chemical structure gives it an advantage over its predecessors. While enzalutamide and apalutamide lose their blocking activity against the F877L mutation and in fact become activators, darolutamide retains its antagonist activity against F877L and T878A due to structural differences in how it binds to the receptor’s docking site.¹²
In patients who have already progressed on other drugs and carry these mutations, darolutamide has shown activity even after the older drugs have undergone an antagonist-agonist switch.
That said, darolutamide is not immune to all resistance mechanisms. The broader problem of cross-resistance among all ARPI agents, mediated in part by AR splice variants such as AR-V7 that bypass the ligand-binding domain entirely, also applies to darolutamide.¹³
Darolutamide is a better-trained guard, but the cancer keeps finding new ways to recruit.
What this means for you
Only 15% of patients with metastatic castration-resistant prostate cancer receive baseline circulating tumor DNA (ctDNA) testing, and only 15% receive more than one test during their course of treatment.¹¹ That means the vast majority of men going through exactly the scenarios described above are doing so without the molecular information that might change the decisions doctors are making on their behalf.
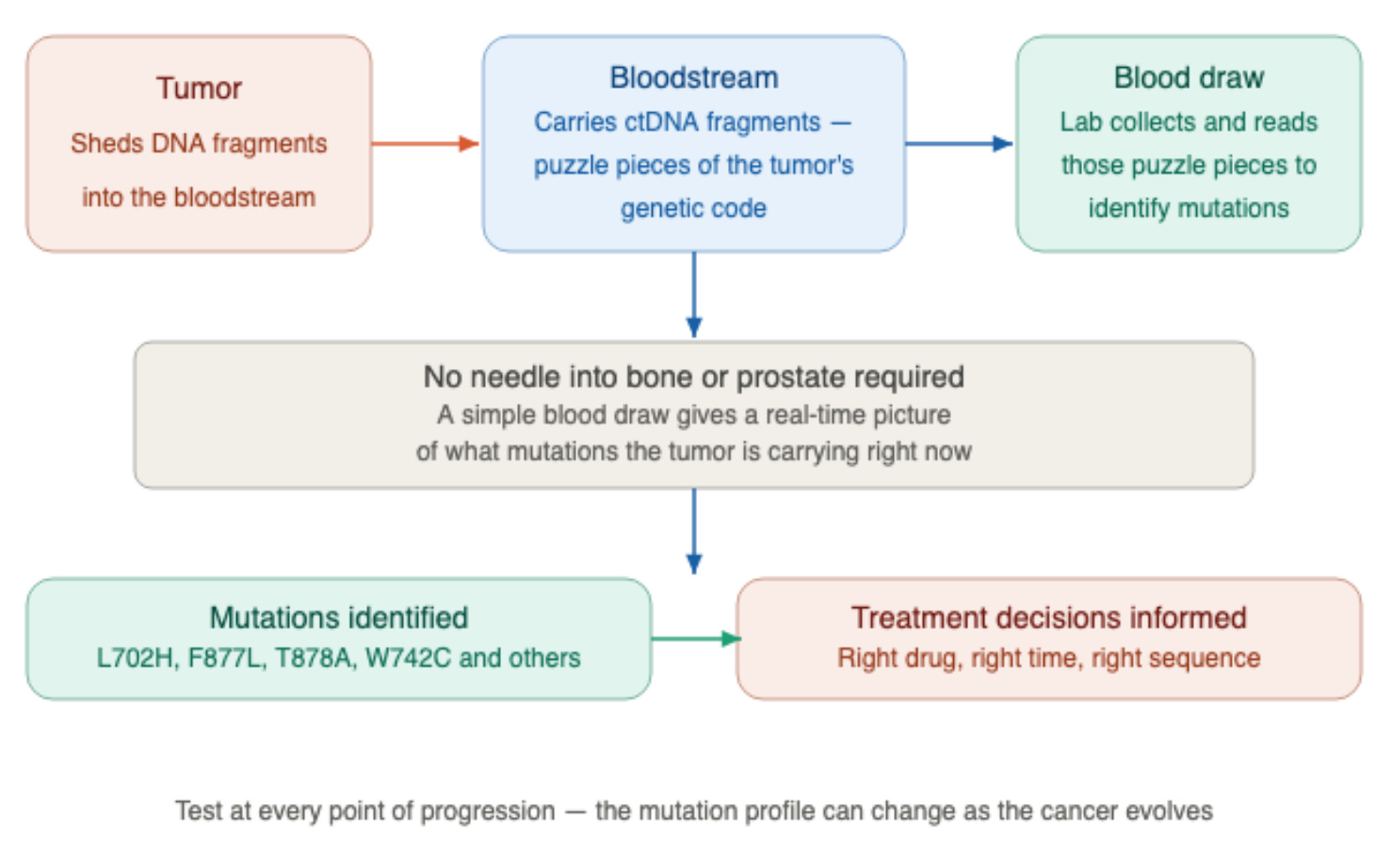
The most concrete action you can take is to ask about circulating tumor DNA (ctDNA) testing, also called a liquid biopsy, at every point of progression.
As a tumor grows, it sheds tiny fragments of its DNA into the bloodstream. Think of them as puzzle pieces of the tumor’s genetic code floating in your blood.
A simple blood draw collects those fragments and reads them, identifying the specific mutations your tumor is carrying right now, without any needle going into bone or the prostate.
The mutation profile can change as the cancer evolves, which is why a single test early in your disease history is not sufficient. You need to know what mutations are present at the moment decisions are being made about your next treatment.
The second action is to understand what the guidelines actually say about switching from one ARPI to another. Both the NCCN and ASCO guidelines are explicit: ARPI switching is not a preferred strategy for patients who progress on ARPIs.⁶˒⁷
After ARPI progression, docetaxel chemotherapy carries a Category 1 (highest level) NCCN recommendation, meaning it is supported by high-quality evidence and uniform expert consensus.⁶
If your doctor is considering moving you from one ARPI to another without molecular testing, that is a conversation worth having.
The PLATO trial data make clear that the benefit of sequential ARPI therapy is modest at best and essentially absent in the reverse direction, and the mutation scenarios described in this article explain why.⁸ Non-AR-directed options include taxane chemotherapy, PARP inhibitors if homologous recombination repair mutations are present, and lutetium-177 PSMA therapy if PSMA expression is retained.⁶˒⁷
The third action is to consider a second opinion at a prostate cancer center of excellence if your PSA is rising through multiple lines of therapy and your doctor hasn’t performed comprehensive molecular testing.
Places like MD Anderson Cancer Center, Memorial Sloan Kettering, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, and Mayo Clinic have molecular tumor boards that meet specifically to work through cases in which standard sequencing logic no longer makes sense.
These cases are complex enough that specialized infrastructure and experience make a real difference. The NCCN acknowledges that “although the optimal sequence of therapies remains undefined, some data are emerging that can help with treatment selection in some cases." ⁶ A prostate center of excellence is where that emerging data is most likely to be applied to your specific situation.
Conclusion
The field of prostate cancer oncology is moving toward strategic, repeated molecular profiling, especially at key inflection points where results could change management. The tests are there, and the science backs them.
What lags behind is consistent clinical application. Until that changes, the burden falls on informed patients to ask the right questions. I hope I’ve provided you enough information to ask some of those questions.
Until the next one, I wish you good health and much love,
Keith
References
Hara T, Miyazaki J, Araki H, et al. Novel mutations of androgen receptor: possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63(1):149–153.
Prekovic S, van Royen ME, Voet AR, et al. The effect of F877L and T878A mutations on androgen receptor response to enzalutamide. Mol Cancer Ther. 2016;15(7):1702–1712.
Antonarakis ES, Zhang N, Saha J, et al. Real-world assessment of AR-LBD mutations in metastatic castration-resistant prostate cancer. J Clin Oncol. 2023.
Attard G, Reid AH, Auchus RJ, et al. Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J Clin Endocrinol Metab. 2012;97(2):507–516.
Li Z, Alyamani M, Li J, et al. Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature. 2016;533(7604):547–551.
National Comprehensive Cancer Network. Prostate Cancer. NCCN Clinical Practice Guidelines in Oncology. Version 1.2026. Updated January 23, 2026.
Garje R, Riaz IB, Naqvi SAA, et al. Systemic therapy in patients with metastatic castration-resistant prostate cancer: ASCO guideline update. J Clin Oncol. 2025.
Khalaf DJ, Annala M, Taavitsainen S, et al. Optimal sequencing of enzalutamide and abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer: a multicentre, randomised, open-label, phase 2, crossover trial. Lancet Oncol. 2019;20(12):1730–1739.
Stewart T, Chandiwana D, Doyle A, et al. Real-world outcomes of patients with metastatic castration-resistant prostate cancer and tumors harboring androgen receptor ligand-binding domain mutations. J Clin Oncol. 2024.
Wyatt AW, Azad AA, Volik SV, et al. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol. 2016;2(12):1598–1606.
Stewart T, Chandiwana D, Doyle A, et al. Real-world outcomes of patients with metastatic castration-resistant prostate cancer and tumors harboring androgen receptor ligand-binding domain mutations. J Clin Oncol. 2024.
Moilanen AM, Riikonen R, Oksala R, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. 2015;5:12007.
Bernstad Salas A, Hofman MS, et al. Cross-resistance among next-generation anti-androgen drugs through the AKR1C3/AR-V7 axis in advanced prostate cancer. Front Oncol. 2021.
Miyazaki J, Nishiyama T, et al. Androgen receptor mutations for precision medicine in prostate cancer. Endocr Relat Cancer. 2022;29(10):R133–R152.
Rathkopf DE, Morris MJ, Fox JJ, et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann Oncol. 2017;28(9):2264–2271.
Rathkopf DE, Morris MJ, Fox JJ, et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann Oncol. 2017;28(9):2264–2271.
Fizazi K, Tran N, Fein L, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med. 2017;377(4):352–360.
Abiraterone-associated mineralocorticoid excess: a case report. Cureus. 2024. PMC10843236.
Yasunaga T, Ruplin A, Fritzsche D, Cheng H. Low-dose abiraterone with a low-fat diet in metastatic prostate cancer. National Community Oncology Dispensing Association (NCODA). Available at: https://www.ncoda.org/news/low-dose-abiraterone-with-a-low-fat-diet-in-metastatic-prostate-cancer/
Disclaimer: The content in this article is for informational and educational purposes only. It does not represent medical advice. Always discuss treatment decisions with your own healthcare provider.
That was some excellent evidence based information. I had a bad feeling regarding bicalutamide long term, and this confirms that for a percentage of patients. It would be interesting to see studies on the effects of Orgovix (relugolix) for 16-24 months after or concommitant IMRT/ HDBT therapy. The side effects were minor, appart from libido
Thank you Dr Keith this an excellent article
I have mCRPC Gleason 10 T4 M1 N1 with NEPC been on Enzalutamide and Regulox for almost 4 years but for the last year my PSA is rising monthly and I am seeking the best way forward can you help